Additional Data
ENHERTU continues to change what is possible with HER2-directed treatment
In 2022, ENHERTU was approved as the first HER2-directed therapy for eligible previously treated patients with HER2-low (IHC 1+ or IHC 2+/ISH–) mBC1,2
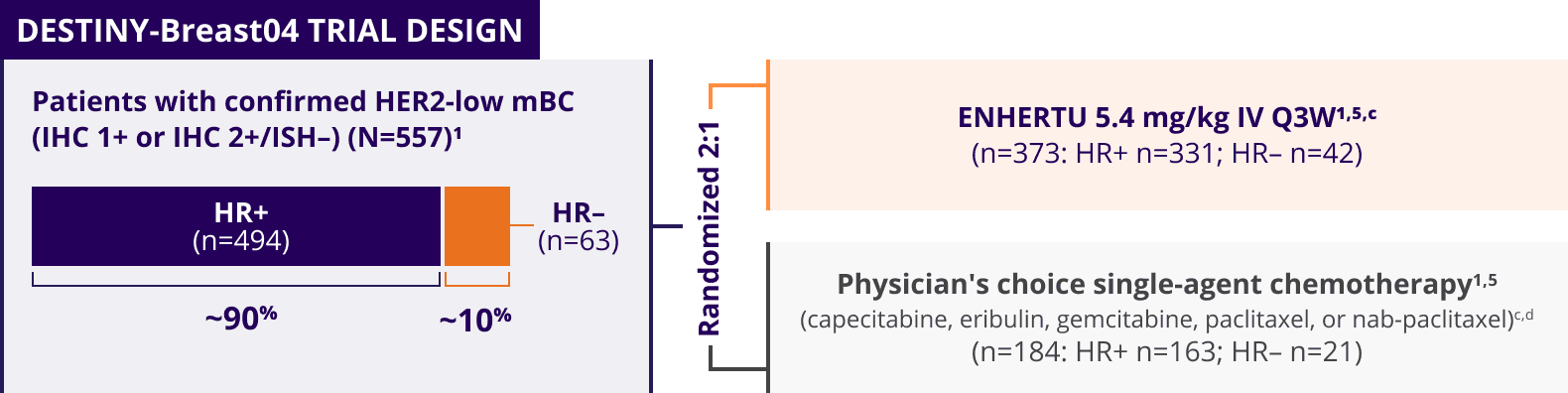
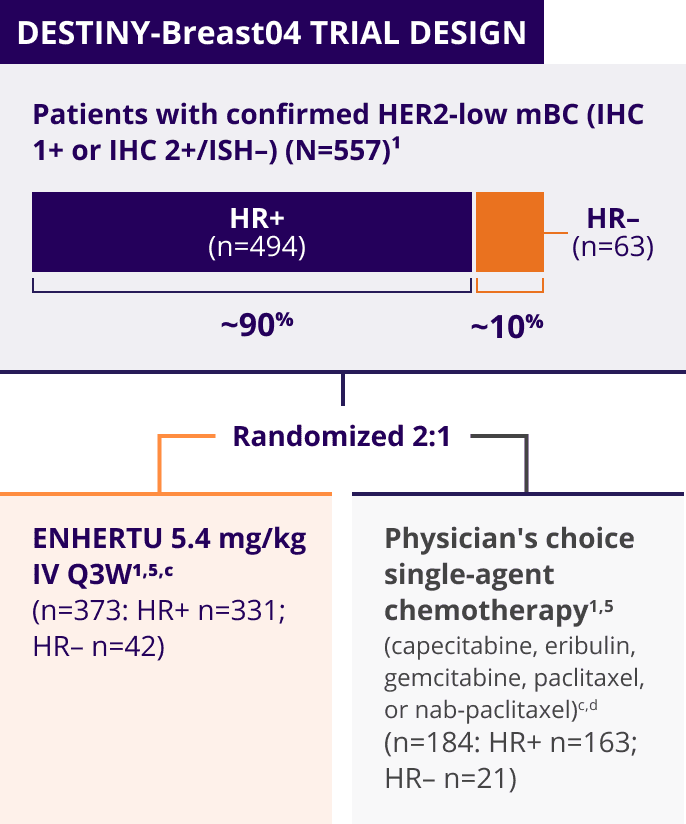
ENHERTU was investigated in DESTINY-Breast04, a landmark, Phase 3, international trial1,3-5
A Phase 3, multicenter, randomized, open-label trial that included patients with1,3,a:
- Unresectable or metastatic HER2-low (HER2 IHC 1+b or IHC 2+/ISH–) breast cancer
- Tumors previously not classified as HER2-positive on any prior pathology testing4
The key efficacy endpoints in DESTINY-Breast04 were tested hierarchically1:
Primary endpoint1
- PFS in the HR+ population, as determined by BICR
Key secondary endpoints1
- PFS (BICR)e in the overall study population (HR+ and HR–)
- OS in the HR+ population
- OS in the overall study population (HR+ and HR–)
Select secondary endpoints3,e,f
- Confirmed ORR and DOR in the HR+ population
- Confirmed ORR and DOR in the overall study population (HR+ and HR–)
Select exploratory endpoints5,6
- PFS, OS, ORR, and DOR in the HR– population
Stratification factors5
- HER2 IHC status (IHC 1+ or IHC 2+/ISH–)
- Number of prior lines of chemotherapy in the metastatic setting (1 or 2)
- HR status/prior CDK4/6 inhibitor (HR+ with prior CDK4/6 inhibitor, HR+ without prior CDK4/6 inhibitor, or HR–)
Patients with HER2-low mBC in DESTINY-Breast04 had 1 or 2 prior lines of chemotherapy in the metastatic setting and, if HR+, were ineligible for or had progressed on endocrine therapy1
aPatients had received no prior HER2-directed therapy.3
bIHC 1+ tumors were ISH– or ISH untested.3
cTreatment was administered until disease progression, death, withdrawal of consent, or unacceptable toxicity.1
d In the control arm, 51.1% of patients received eribulin, 20.1% received capecitabine, 10.3% received nab-paclitaxel, 10.3% received gemcitabine, and 8.2% received paclitaxel.5
eAccording to mRECIST v1.1.3
fBoth BICR and investigator assessment were performed.3
DESTINY-Breast04 evaluated patients with HER2-low mBC (IHC 1+ and IHC 2+/ISH–)1
Select baseline patient demographics and clinical characteristics1,6,g,h
|
DESTINY-Breast04
(N=557) |
||
|---|---|---|
| Age, median | 57 years (range: 28-81) | |
| Hormone receptor status | HR+ | 89% |
| HR– | 11% | |
| HER2 IHC score | 1+ | 58% |
| 2+/ISH– | 42% | |
| Number of prior lines of endocrine therapy in the metastatic setting in HR+/HER2-low mBC patients (n=494) | 0 | 9% |
| 1 | 31% | |
| 2 | 33% | |
| ≥3 | 27% | |
| Prior CDK4/6 inhibitor in HR+/HER2-low mBC patients (n=494) | 70% | |
|
Number of prior chemotherapy regimens in the metastatic setting |
1 | 58% |
| 2 | 41% | |
| Prior lines of systemic therapy in the metastatic setting, mediani | 3 (range: 1-9) | |
| Sites of metastases | Liver metastases | 70% |
| Lung metastases | 33% | |
| Brain metastases j | 6% | |
|
DESTINY-Breast04
(N=557) |
|
|---|---|
| Age, median |
57 years (range: 28-81) |
| Hormone receptor status | |
| HR+ | 89% |
| HR– | 11% |
| HER2 IHC score | |
| 1+ | 58% |
| 2+/ISH– | 42% |
| Number of prior lines of endocrine therapy in the metastatic setting in HR+/HER2-low mBC patients (n=494) | |
| 0 | 9% |
| 1 | 31% |
| 2 | 33% |
| ≥3 | 27% |
| Prior CDK4/6 inhibitor in HR+/HER2-low mBC patients (n=494) | 70% |
|
Number of prior chemotherapy regimens in the metastatic setting |
|
| 1 | 58% |
| 2 | 41% |
| Prior lines of systemic therapy in the metastatic setting, mediani | 3 (range: 1-9) |
| Sites of metastases | |
| Liver metastases | 70% |
| Lung metastases | 33% |
| Brain metastases j | 6% |
- Gender: 99.6% were female, 0.4% were male1
- Race/ethnicity: 48% were White, 40% were Asian, 3.8% were Hispanic/Latino, and 2% were Black or African American1
- ECOG PS: 55% had ECOG PS 0 and 45% had ECOG PS 1 at baseline1
- In the HR+ cohort, median prior lines of ET: 2 (range: 0-9)1
- Early progressors (progression in the neo/adjuvant setting): 3.9%1
gPatients with HR-negative mBC were not included in the primary efficacy analysis.5
hIn some instances, percentages do not add up to 100% due to rounding.
iIn the overall study population (N=557), 10% of patients had received 1 prior line of systemic therapy in the metastatic setting, 27% had received 2 prior lines, and 62% had received ≥3 prior lines.5
jPatients with treated brain metastases who were no longer symptomatic and who required no treatment with corticosteroids or anticonvulsants could be included in the study if they had recovered from the acute toxic effect of radiotherapy. A minimum of 2 weeks must have elapsed between the end of whole brain radiotherapy and study enrollment.3
Summary of DESTINY-Breast04 efficacy results by patient population1,5,6,k,l
|
Overall study population (N=557) |
HR+/HER2-low cohort (n=494) |
Exploratory HR– /HER2-low cohort (n=58) |
||||
|---|---|---|---|---|---|---|
|
ENHERTU (n=373) |
Chemo- therapy (n=184) |
ENHERTU (n=331) |
Chemo- therapy (n=163) |
ENHERTU (n=40) |
Chemo- therapy (n=18) |
|
|
mPFS (mo) (95% Cl) |
9.9 (9.0, 11.3) |
5.1 (4.2, 6.8) |
10.1 (9.5, 11.5) |
5.4 (4.4, 7.1) |
8.5 (4.3, 11.7) |
2.9 (1.4, 5.1) |
| HR (95% CI; P-value) |
0.50 (0.40, 0.63; P<0.0001) |
0.51 (0.40, 0.64; P<0.0001) |
0.46 (0.24, 0.89) |
|||
|
mOS (mo) (95% Cl) |
23.4 (20.0, 24.8) |
16.8 (14.5, 20.0) |
23.9 (20.8, 24.8) |
17.5 (15.2, 22.4) |
18.2 (13.6, NE) |
8.3 (5.6, 20.6) |
| HR (95% CI; P-value) |
0.64 (0.49, 0.84; P=0.001) |
0.64 (0.48, 0.86; P=0.0028) |
0.48 (0.24, 0.95) |
|||
|
ORR (n; 95% CI) |
52.3% (195; 47.1, 57.4) |
16.3% (30; 11.3, 22.5) |
52.9% (175; 47.3, 58.4) |
16.6% (27; 11.2, 23.2) |
50.0% (20; 33.8, 66.2) |
16.7% (3; 3.6, 41.4) |
| CR |
3.5% (n=13) |
1.1% (n=2) |
3.6% (n=12) |
0.6% (n=1) |
2.5% (n=1) |
5.6% (n=1) |
| PR |
49.1% (n=183) |
15.2% (n=28) |
49.5% (n=164) |
16.0% (n=26) |
47.5% (n=19) |
11.1% (n=2) |
|
mDOR (mo) (95% Cl) |
10.7 (8.5, 13.2) |
6.8 (6.0, 9.9) |
10.7 (8.5, 13.7) |
6.8 (6.5, 9.9) |
8.6 (7.1, 13.9) |
4.9 (3.7, 6.0) |
|
Overall study population (N=557) |
||
|---|---|---|
|
ENHERTU (n=373) |
Chemo- therapy (n=184) |
|
|
mPFS (mo) (95% Cl) |
9.9 (9.0, 11.3) |
5.1 (4.2, 6.8) |
|
HR (95% CI; P-value) |
0.50 (0.40, 0.63; P<0.0001) |
|
|
mOS (mo) (95% Cl) |
23.4 (20.0, 24.8) |
16.8 (14.5, 20.0) |
|
HR (95% CI; P-value) |
0.64 (0.49, 0.84; P=0.001) |
|
|
ORR (n; 95% CI) |
52.3% (195; 47.1, 57.4) |
16.3% (30; 11.3, 22.5) |
| CR |
3.5% (n=13) |
1.1% (n=2) |
| PR |
49.1% (n=183) |
15.2% (n=28) |
|
mDOR (mo) (95% Cl) |
10.7 (8.5, 13.2) |
6.8 (6.0, 9.9) |
|
HR+/HER2-low cohort (n=494) |
||
|
ENHERTU (n=331) |
Chemo- therapy (n=163) |
|
|
mPFS (mo) (95% Cl) |
10.1 (9.5, 11.5) |
5.4 (4.4, 7.1) |
|
HR (95% CI; P-value) |
0.51 (0.40, 0.64; P<0.0001) |
|
|
mOS (mo) (95% Cl) |
23.9 (20.8, 24.8) |
17.5 (15.2, 22.4) |
|
HR (95% CI; P-value) |
0.64 (0.48, 0.86; P=0.0028) |
|
|
ORR (n; 95% CI) |
52.9% (175; 47.3, 58.4) |
16.6% (27; 11.2, 23.2) |
| CR |
3.6% (n=12) |
0.6% (n=1) |
| PR |
49.5% (n=164) |
16.0% (n=26) |
|
mDOR (mo) (95% Cl) |
10.7 (8.5, 13.7) |
6.8 (6.5, 9.9) |
|
Exploratory HR–/HER2-low cohort (n=58) |
||
|
ENHERTU (n=40) |
Chemo- therapy (n=18) |
|
|
mPFS (mo) (95% Cl) |
8.5 (4.3, 11.7) |
2.9 (1.4, 5.1) |
|
HR (95% CI; P-value) |
0.46 (0.24, 0.89) |
|
|
mOS (mo) (95% Cl) |
18.2 (13.6, NE) |
8.3 (5.6, 20.6) |
|
HR (95% CI; P-value) |
0.48 (0.24, 0.95) |
|
|
ORR (n; 95% CI) |
50.0% (20; 33.8, 66.2) |
16.7% (3; 3.6, 41.4) |
| CR |
2.5% (n=1) |
5.6% (n=1) |
| PR |
47.5% (n=19) |
11.1% (n=2) |
|
mDOR (mo) (95% Cl) |
8.6 (7.1, 13.9) |
4.9 (3.7, 6.0) |
kFor the primary and secondary endpoints, the hormone receptor status is based on data collected using the interactive web/voice response system at the time of randomization, which includes mis-stratified patients.5
lFor other endpoints, hormone receptor status is based on data from the electronic data capture corrected for mis-stratification.5
- The HR– cohort was an exploratory population. The data are descriptive and were not tested for statistical significance, nor powered to show a difference between treatment arms. Therefore, the clinical significance of these data is not known
- ORR and mDOR were not tested for statistical significance and were not powered to show differences between treatment arms. Therefore, the clinical significance of these data is not known
An established benefit-risk profile with ENHERTU in DESTINY-Breast04
The majority of adverse reactions were Grade 1 or 21
- The median duration of treatment was 8 months (range: 0.2-33) with ENHERTU and 3.5 months (range: 0.3-17.6) with chemotherapy1,5
Common adverse reactions (≥10% all Grades or ≥2% Grades 3 or 4) in patients treated with ENHERTU in DESTINY-Breast041
| Adverse reactions |
ENHERTU 5.4
mg/kg (n=371) |
Chemotherapy (n=172) |
|||
|---|---|---|---|---|---|
|
All Grades (%) |
Grades 3–4 (%) |
All Grades (%) |
Grades 3–4 (%) |
||
|
Gastrointestinal disorders |
Nausea | 76 | 4.6 | 30 | 0 |
| Vomiting | 40 | 1.6 | 13 | 0 | |
| Constipation | 34 | 0.8 | 22 | 0 | |
| Diarrhea | 27 | 1.3 | 22 | 1.7 | |
| Abdominal painm | 18 | 0.5 | 13 | 0 | |
| Stomatitisn | 13 | 0.3 | 12 | 0.6 | |
|
General disorders and administration site conditions |
Fatigueo | 54 | 9 | 48 | 4.7 |
| Pyrexia | 12 | 0.3 | 13 | 0 | |
|
Skin and subcutaneous tissue disorders |
Alopecia | 40 | 0 | 33 | 0 |
| Rashp | 13 | 0 | 23 | 4.7 | |
|
Metabolism and nutrition disorders |
Decreased appetite | 32 | 2.4 | 19 | 1.2 |
|
Musculoskeletal and connective tissue disorders |
Musculoskeletal painq | 32 | 1.3 | 31 | 0.6 |
| Investigations | Decreased weight | 16 | 0.3 | 8 | 0 |
| Vascular disorders | Hemorrhager | 16 | 0 | 3.5 | 0 |
| Nervous system disorders | Headaches | 15 | 0.3 | 6 | 0 |
| Peripheral neuropathyt | 13 | 0 | 29 | 5 | |
| Dizzinessu | 11 | 0.5 | 6 | 0 | |
| Infections and infestations | Upper respiratory tract infectionv | 14 | 0.3 | 5 | 0 |
|
Respiratory, thoracic, and mediastinal disorders |
Interstitial lung diseasew | 12 | 1.3 | 0.6 | 0 |
| Dyspnea | 10 | 1.3 | 9 | 1.2 | |
|
Adverse reactions |
ENHERTU 5.4 mg/kg (n=371) |
Chemo- therapy (n=172) |
||
|---|---|---|---|---|
|
All Gra- des (%) |
Gra- des 3–4 (%) |
All Gra- des (%) |
Gra- des 3–4 (%) |
|
| Gastrointestinal disorders | ||||
| Nausea | 76 | 4.6 | 30 | 0 |
| Vomiting | 40 | 1.6 | 13 | 0 |
| Constipation | 34 | 0.8 | 22 | 0 |
| Diarrhea | 27 | 1.3 | 22 | 1.7 |
| Abdominal painm | 18 | 0.5 | 13 | 0 |
| Stomatitisn | 13 | 0.3 | 12 | 0.6 |
| General disorders and administration site conditions | ||||
| Fatigueo | 54 | 9 | 48 | 4.7 |
| Pyrexia | 12 | 0.3 | 13 | 0 |
| Skin and subcutaneous tissue disorders | ||||
| Alopecia | 40 | 0 | 33 | 0 |
| Rashp | 13 | 0 | 23 | 4.7 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 32 | 2.4 | 19 | 1.2 |
| Musculoskeletal and connective tissue disorders | ||||
| Musculo-skeletal painq | 32 | 1.3 | 31 | 0.6 |
| Investigations | ||||
| Decreased weight | 16 | 0.3 | 8 | 0 |
| Vascular disorders | ||||
| Hemorrhager | 16 | 0 | 3.5 | 0 |
| Nervous system disorders | ||||
| Headaches | 15 | 0.3 | 6 | 0 |
| Peripheral neuropathyt | 13 | 0 | 29 | 5 |
| Dizzinessu | 11 | 0.5 | 6 | 0 |
| Infections and infestations | ||||
| Upper respiratory tract infectionv | 14 | 0.3 | 5 | 0 |
| Respiratory, thoracic, and mediastinal disorders | ||||
| Interstitial lung diseasew | 12 | 1.3 | 0.6 | 0 |
| Dyspnea | 10 | 1.3 | 9 | 1.2 |
mIncluding abdominal pain, abdominal discomfort, lower abdominal pain, and upper abdominal pain.
nIncluding stomatitis, aphthous ulcer, mouth ulceration, and pharyngeal inflammation.
oIncluding fatigue, asthenia, and malaise.
pIncluding rash, pustular rash, pruritic rash, maculo-papular rash, palmar-plantar erythrodysesthesia syndrome, papular rash, macular rash, eczema, erythema multiforme, dermatitis, urticarial dermatitis, drug eruption, and dermatitis bullous.
qIncluding back pain, myalgia, pain in extremity, musculoskeletal pain, bone pain, musculoskeletal chest pain, arthralgia, noncardiac chest pain, musculoskeletal stiffness, arthritis, spinal pain, and neck pain.
rIncluding esophageal varices, hemorrhage, hemorrhoidal hemorrhage, epistaxis, hematuria, conjunctival hemorrhage, vaginal hemorrhage, gingival bleeding, genital hemorrhage, eye hemorrhage, hemoptysis, hemorrhagic cystitis, pharyngeal hemorrhage, rectal hemorrhage, upper gastrointestinal hemorrhage, and esophageal hemorrhage.
sIncluding headache and migraine.
tIncluding peripheral neuropathy, peripheral sensory neuropathy, peripheral motor neuropathy, polyneuropathy, paresthesia, hypoesthesia, dysesthesia, and neuralgia.
uIncluding dizziness, postural dizziness, and vertigo.
vIncluding upper respiratory tract infection, influenza, influenza-like illness, nasopharyngitis, pharyngitis, sinusitis, and rhinitis.
wInterstitial lung disease includes events that were adjudicated as drug-induced ILD for ENHERTU: interstitial lung disease, pneumonitis, organizing pneumonia, pneumonia, and radiation pneumonitis.
Adverse reactions may require dose modifications1
|
Clinically relevant AR considerations |
ENHERTU 5.4 mg/kg (n=371) |
Chemo-
therapy
(n=172) |
|
|---|---|---|---|
| Serious adverse reactions1,5,6 | 28% | 25% |
|
| Discontinuations due to adverse reactions1,5 | 16% | 8% |
|
| Dose interruptions due to adverse reactions1,4,6 | 39% | 42% |
|
| Dose reductions due to adverse reactions1,5,6 | 23% | 38% |
|
|
Clinically relevant AR considerations |
|
| Serious adverse reactions1,5,6 | |
|
ENHERTU 5.4 mg/kg (n=371) |
28% |
|
Chemotherapy
(n=172) |
25% |
|
|
|
|
|
Clinically relevant AR considerations |
|
|
Discontinuations due to adverse reactions1,5 |
|
|
ENHERTU 5.4 mg/kg (n=371) |
16% |
|
Chemotherapy (n=172) |
8% |
|
|
|
|
|
Clinically relevant AR considerations |
|
|
Dose interruptions due to adverse reactions1,4,6 |
|
|
ENHERTU 5.4 mg/kg (n=371) |
39% |
|
Chemotherapy (n=172) |
42% |
|
|
|
|
|
Clinically relevant AR considerations |
|
|
Dose reductions due to adverse reactions1,5,6 |
|
|
ENHERTU 5.4 mg/kg (n=371) |
23% |
|
Chemotherapy (n=172) |
38% |
|
|
|
|
- Other clinically relevant adverse reactions reported in ≤10% of patients treated with ENHERTU were cough (10%), dysgeusia (10%), abdominal distension (5%), blurred vision (4.9%), pruritus (3.2%), gastritis (2.7%), skin hyperpigmentation (2.7%), flatulence (2.4%), dehydration (1.9%), febrile neutropenia (1.1%), and infusion-related reactions (0.5%)1
In DESTINY-Breast04, Grade 5 ILD/pneumonitis events were observed in 0.8% of patients (n=3/371)1,5,x
- Of the 371 patients treated with ENHERTU 5.4 mg/kg, ILD occurred in 12.1% of patients (n=45/371)5
xGrade 5=fatal cases.
AR, adverse reaction; BICR, blinded independent central review; CDK4/6, cyclin-dependent kinases 4 and 6; CI, confidence interval; CR, complete response; DOR, duration of response; ECOG, Eastern Cooperative Oncology Group; ET, endocrine therapy; HER2, human epidermal growth factor receptor 2; HR, hazard ratio; HR–, hormone receptor-negative; HR+, hormone receptor-positive; IHC, immunohistochemistry; ILD, interstitial lung disease; ISH, in situ hybridization; IV, intravenous; mBC, metastatic breast cancer; mDOR, median duration of response; mOS, median overall survival; mPFS, median progression-free survival; mRECIST, modified Response Evaluation Criteria in Solid Tumors; NE, not evaluable; ORR, objective response rate; OS, overall survival; PFS, progression-free survival; PR, partial response; PS, performance score; Q3W, every 3 weeks.